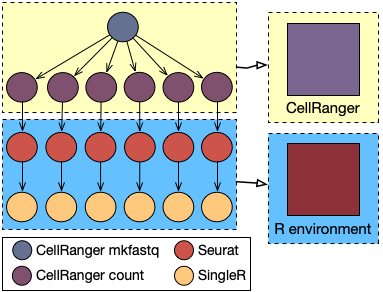
The single-cell RNA sequencing (scRNA-seq) analysis technique is essential to assess fundamental biological properties of cells populations and biological systems at unprecedented resolution. Identifying subpopulations of cells in scRNA-seq experiments is one of the most frequently performed analysis of single-cell data. Subpopulation discovery commonly relies on clustering algorithms, e.g. the Seurat R package (see also this post).
The rCASC library
The rCASC library is specifically designed to provide an integrated analysis environment for cell subpopulation discovery, providing high flexibility and enabling computation reproducibility. In detail, rCASC supports three different analysis steps: raw data preprocessing, subpopulation discovery via clustering, and cluster-specific gene signature detection.
These three analysis steps require different computational resources and computing models. They are therefore suitable to be described as a hybrid workflow running on a heterogeneous computing environment, composed of CPU and GPU architectures, multi-core machines and multi-server deployments. In particular, clustering is the most computationally demanding activity.
StreamFlow application
The StreamFlow framework has been leveraged to execute three different clustering algorithms (SIMLR, Griph, and tSne) running on up to 8 virtual machines (8 cores, 32 GB RAM each) allocated on the HPC4AI Cloud facility at Università di Torino. In particular, the goal was to measure the speedup achievable using StreamFlow with respect to a single multi-core server.
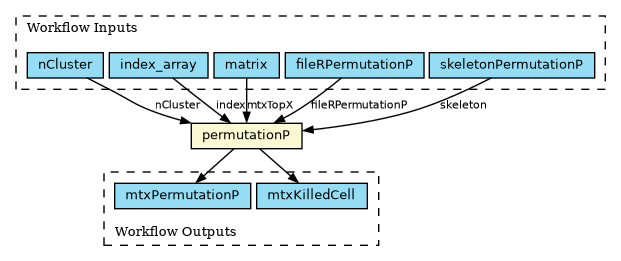
A single-step workflow running a subpopulation clustering analysis has been implemented in the Common Workflow Language (CWL) format, as shown in the figure above. Implementing it as a workflow allows using the CWL scatter feature to distribute independent portions of the workload across multiple locations for concurrent execution. In this case, the scatter is executed on the index_array input field. The code is available on GitHub.
All three clustering algorithms showed a significant speedup with the progressive increase of compute nodes. In particular, on 8 nodes Griph and SIMLR obtained good speedup values of 4x and 4.7x, respectively. Also the tSne algorithm obtained a still significant speedup of 2.5x, supporting the general usefulness of hybrid workflows in subpopulation clustering analyses.
S. G. Contaldo, L. Alessandri, I. Colonnelli, M. Beccuti, and M. Aldinucci, “Bringing cell subpopulation discovery on a cloud-HPC using rCASC and StreamFlow,” in Single cell transcriptomics: methods and protocols, R. A. Calogero and V. Benes, Eds., New York, NY: Springer US, 2023, p. 337–345. doi: 10.1007/978-1-0716-2756-3_17.