The Federated Learning (FL) approach is a paradigmatic example of modern AI applications. FL tackles the problem of collaboratively training a Machine Learning model using distributed data silos, where data cannot leave the owner’s infrastructure to ensure privacy and secrecy. Modelling a FL workflow is challenging because it requires federating infrastructures and iterative execution patterns.
Existing FL architectures
The typical runtime architecture of FL frameworks (e.g., Intel OpenFL and Flower) is a master/worker. Each worker is deployed onto a different silo, where it trains a private copy of a Deep Neural Network (DNN). At the end of each training round, each worker sends its model to the master, which computes an aggregated model using a configurable algorithm and broadcasts it back to workers for the next round.
Some recent FL frameworks drop the constraint of a single centralized aggregator, either relying on a tree-based infrastructure or implementing a fully decentralized peer-to-peer aggregation protocol. However, the communication topology is always an intrinsic characteristic of the framework implementation.
In research scenarios, data providers are usually independent entities with heterogeneous data treatment protocols, storage infrastructures, and access policies. Therefore, cross-silo FL pipelines are perfect candidates to be modeled as hybrid workflows.
StreamFlow application
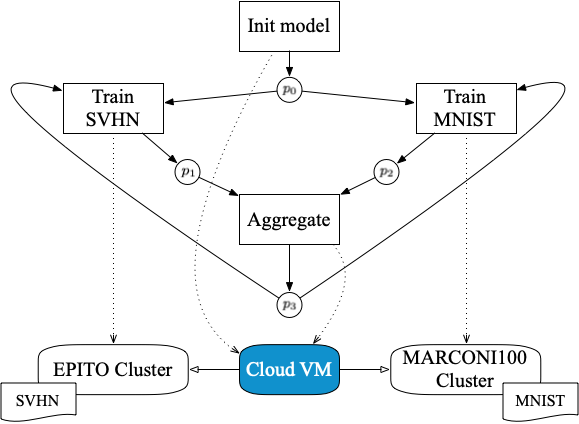
StreamFlow has been used to execute a cross-cluster FL pipeline, where two independent HPC clusters train a model on two different private datasets and a Cloud VM acts as a centralized aggregator.
As a first step, a Common Workflow Language (CWL) description of a FL pipeline has been designed. The pipeline trains a VGG16 DNN over two datasets: a standard MNIST residing on the CINECA MARCONI100 HPC facility (2×16-core IBM POWER9 AC922, 256 GB RAM, and 4 NVIDIA V100 GPUs per node), and a grayscaled version of SVHN residing in the EPITO bare metal partition of the HPC4AI facility at Università di Torino (80-core Arm Neoverse-N1, 512GB RAM, and 2 NVIDIA A100 GPU per node). Note that, up to version v1.2, CWL does not support iterative constructs. However, this pipeline is the first real case iterative CWL workflow, relying on the recently proposed Loop extension. The code is available on GitHub.
Two different FL configurations have been tested: 100 rounds of 1 epoch each and 50 rounds of 2 epochs each, using the well known Federated Averaging (FedAvg) algorithm. Note that the typical master/worker architecture of FL frameworks requires direct bidirectional communications between the aggregator and each worker node. This is not compatible with the typical network configuration of an HPC facility, where worker nodes cannot open outbound connections. Therefore, StreamFlow is a key enabling technology for cross-cluster FL.
To compare performances with a baseline, the pipeline has also been tested on a pure cloud execution environment, replacing the two clusters with two VMs (8 cores, 32 GB RAM, and 1 NVIDIA T5 GPU each) running on the cloud partition of the HPC4AI facility. The performance obtained with the StreamFlow execution of the pipeline has been compared with an equivalent training workload managed by the Intel OpenFL framework. Collected results are comparable in terms of both accuracy and time-to-solution, showing how general-purpose hybrid workflows are ready to provide adequate performance in the FL field.
I. Colonnelli, B. Casella, G. Mittone, Y. Arfat, B. Cantalupo, R. Esposito, A. R. Martinelli, D. Medić, and M. Aldinucci, “Federated Learning meets HPC and cloud,” in Astrophysics and Space Science Proceedings, Catania, Italy, 2023, p. 193–199. doi:10.1007/978-3-031-34167-0_39