The idea behind novel single-cell RNA sequencing (scRNA-seq) pipelines is to isolate single cells through microfluidic approaches and generating sequencing libraries in which the transcripts are tagged to track their cell of origin. Modern scRNA-seq platforms are capable of analysing from 500 to 20,000 cells in each run. Then, combined with massive high-throughput sequencing producing billions of reads, scRNA-seq allows the assessment of fundamental biological properties of cells populations and biological systems at unprecedented resolution.
Single-cell pipeline
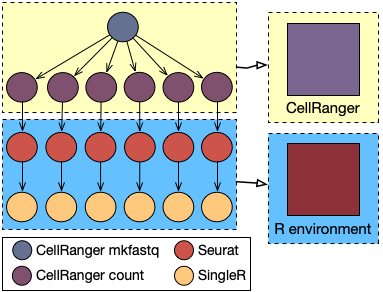
A typical pipeline for single-cell transcriptomic data analysis can be broadly divided into two main parts: the creation of the count matrix, performed according to the adopted single-cell experimental technology and the used sequencing approach, and its statistical analysis, usually using ad-hoc developed software in Python or R.
Considering a typical 10x Genomics experiment followed by an Illumina Novaseq sequencing, the first part of the pipeline will be performed using a tool called CellRanger. In particular, this part of the analysis will consist in two steps: the creation of the fastq files (the raw sequences of the four bases) from the flowcell provided in output by the sequencer and the alignment of the reads against the reference genome, in order to find for each gene how many reads have been captured.
The Seurat R package is then used to load data into the R environment and to perform some preliminary operations (such as outlier filtering, normalisation and dimensionality reduction) and clustering, identifying marker genes for each cluster by comparing the expression profile of the cells inside the cluster with all the other cells (see also this post).
Finally, the SingleR package is used to identify the type of each cell (such as Blood Cell, Bone Cell, and Stem Cell) in an unbiased way, leveraging reference transcriptomic datasets of pure cell types to infer the identity of every single cell independently.
The first two steps of the pipeline, related to the creation of the count matrix, have much higher requirements in terms of computing power. Typically, a significant speedup can be appreciated until up to 32 cores and 128GB of memory. Conversely, R packages are not able to fully exploit a such high level of parallelism, resulting in a waste of HPC resources.
StreamFlow application
In such context, StreamFlow has been leveraged to execute the workflow on top of an hybrid cloud-HPC environment without modifying the original codebase. In particular, CellRanger computations have been performed on the C3S HPC facility at Università di Torino, while the remaining steps have been offloaded to a Kubernetes instance running on top of the GARR cloud infrastructure.
The total execution time of the workflow on top of such hybrid infrastructure is comparable with a full-HPC execution, demonstrating how the StreamFlow approach can be beneficial to obtain a more efficient resource allocation without significant performance drops.
I. Colonnelli, B. Cantalupo, I. Merelli and M. Aldinucci, “StreamFlow: cross-breeding cloud with HPC,” in IEEE Transactions on Emerging Topics in Computing, vol. 9, iss. 4, p. 1723-1737, 2021. doi: 10.1109/TETC.2020.3019202.